DNAmixtures is a statistical framework for analysis of DNA samples from one or multiple donors. It implements the statistical model introduced in Analysis of Forensic DNA mixtures with Artefacts (published with discussion in Journal of the Royal Statistical Society, series C).
DNAmixtures has been used to give evidence in several cases in both Denmark and the United Kingdom. Contact Therese Graversen for inquiries related to the use of DNAmixtures in criminal casework.
- Common forensic applications
- Computation of likelihood ratios
- Deconvolution of the DNA mixture
- Combined analysis of multiple traces of DNA
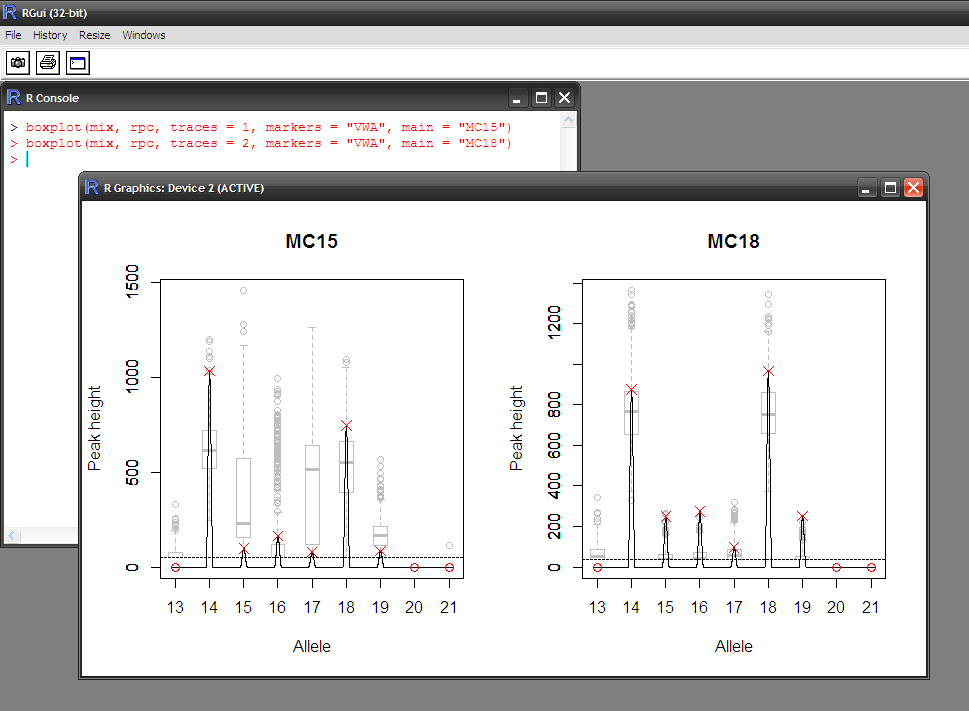
- Visual assessment of the statistical modelling
- Does the hypothesis explain the data well?
- Is the distribution of peak heights adequate?
- Efficient exact computation
- Runs on a standard desktop or laptop
- Allows a higher number of contributors
- A complete statistical framework
- Exact computation of the likelihood function
- Maximum likelihood estimation of parameters
- Simulation of genotypes and peak heights
- Access to various conditional distributions of peak heights given data
System requirements
DNAmixtures may be installed on any system (Windows, Linux, or MacOS), on which the following three programs are installed:
| R | Statistical software |
|---|---|
| Hugin | Bayesian network software |
| RHugin | API for calling Hugin from within R |
For an installation guide, please see the installation page.
Now available: Hugin-free lite-version of DNAmixtures
The DNAmixturesLite package is intended as a service to enable users to try DNAmixtures without purchasing a commercial licence for Hugin. When at all possible, we strongly recommend the use of DNAmixtures rather than its lite-version. While the lite-version seeks to provide the full functionality of DNAmixtures, note that computations are much less efficient and that there are some differences in available functionality.
DNAmixturesLiteQuestions and comments are very welcome and can be directed to the author, Therese Graversen.
